Run PyXtal executables
Currently, we provide several utilities to the users so that they can run the code from command line with Python scripting. They include:
Pyxtal_main.py: a tool to generate atomic/molecular crystalsPyxtal_symmetry.py: a tool to access the symmetry information
After a successfull installation, all of them can be
accessed by invoking the -h command:
$ pyxtal_main.py -h
______ _ _ _
(_____ \ \ \ / / | |
_____) ) _ \ \/ / |_ ____| |
| ____/ | | | ) (| _)/ _ | |
| | | |_| |/ /\ \ |_( (_| | |___
|_| \__ /_/ \_\___)__|_|_____)
(____/
----------------------(version 0.1.4 )----------------------
A Python package for random crystal generation
The source code is available at https://github.com/MaterSim/pyxtal
Developed by Zhu's group at University of Nevada Las Vegas
usage: pyxtal_main.py [-h] [-s sg] [-e element] [-n numIons] [-f factor]
[-a attempts] [-o outdir] [-d dimension] [-t thickness]
[-m]
optional arguments:
-h, --help show this help message and exit
-s sg, --symmetry sg desired symmetry, number or string, e.g., 36, Pbca, Ih
-e element, --element element
desired elements: e.g., Li
-n numIons, --numIons numIons/numMols
desired numbers of atoms: 16
-f factor, --factor factor
volume factor: default 1.0
-a attempts, --attempts attempts
number of crystals to generate: default 1
-o outdir, --outdir outdir
Directory for storing output cif files: default 'out'
-d dimension, --dimension dimension
desired dimension: (3, 2, 1, 0): default 3
-t thickness, --thickness thickness
Thickness of a 2D crystal, or area of a 1D crystal,
None generates a value automatically: default None
-m, --molecular molecular? default: False
PyXtal_symmetry utility
PyXtal_symmetry.py is a utility to handle the generation of moelcular crystals.
-d, the dimension, e.g.,
3,2,1,0. The defult is3.-s: the target symmetry, either by string (e.g.,
Ih,Pbca) and integer (61).
$ pyxtal_symmetry.py -s 36
-- Space group # 36 (Cmc2_1)--
8b site symm: 1
x, y, z
-x, -y, z+1/2
x, -y, z+1/2
-x, y, z
x+1/2, y+1/2, z
-x+1/2, -y+1/2, z+1/2
x+1/2, -y+1/2, z+1/2
-x+1/2, y+1/2, z
4a site symm: m..
0, y, z
0, -y, z+1/2
1/2, y+1/2, z
1/2, -y+1/2, z+1/2
$ pyxtal_symmetry.py -s 20 -d 2
-- Layer group # 20 (p2_122)--
4d site symm: 1
x, y, z
x+1/2, -y, -z
-x+1/2, y, -z
-x, -y, z
2c site symm: .2.
1/4, y, 0
3/4, -y, 0
2b site symm: ..2
0, 1/2, z
1/2, 1/2, -z
2a site symm: ..2
0, 0, z
1/2, 0, -z
if the -s tag is not given, it will output the list of all possible symmetry
groups for the given dimension.
$ pyxtal_symmetry.py -d 3
space_group
1 P1
2 P-1
3 P2
4 P2_1
5 C2
6 Pm
7 Pc
8 Cm
9 Cc
10 P2/m
11 P2_1/m
12 C2/m
13 P2/c
14 P2_1/c
15 C2/c
16 P222
17 P222_1
18 P2_12_12
19 P2_12_12_1
20 C222_1
...
...
212 P4332
213 P4_132
214 I4_132
215 P-43m
216 F-43m
217 I-43m
218 P-43n
219 F-43c
220 I-43d
221 Pm-3m
222 Pn-3n
223 Pm-3n
224 Pn-3m
225 Fm-3m
226 Fm-3c
227 Fd-3m
228 Fd-3c
229 Im-3m
230 Ia-3d
PyXtal_main utility
PyXtal_main.py is a utility to handle the generation of atomic crystals.
Typically, four arguments are requried to describe the target structure:
-d, the dimension, e.g.,
3,2,1,0.-s: the target symmetry (space, layer, rod, point group information), either by string (e.g.,
Ih,Pbca) and integer (61).-e: the list of elements, e.g.,
Si,Si, O-n: the number of atoms in the target primitive unit cell, e.g.,
12,4, 8. The size should be consistent with the-etag.
For group setting, please refer to the Group Setting page. To our knowledge, PyXtal is perhaps the only open source code which can handle the crystal symmetry generation from 0 to 3 dimensional systems. Below we will introduce its capability in detail.
A quick example of C60
Below is a quick example to generate a random C60 clusters with icosahedral
(Ih) symmetry.
$ pyxtal_main.py -e C -n 60 -d 0 -s Ih
Symmetry requested: 56(Ih), generated: Ih
Output to out/C60.xyz
As described in the screen output, the run will generate a file called
out/C60.xyz which stores the structural information about C60.
One can thus visualize via different third-party packages. For instance,
below is the output from VESTA.
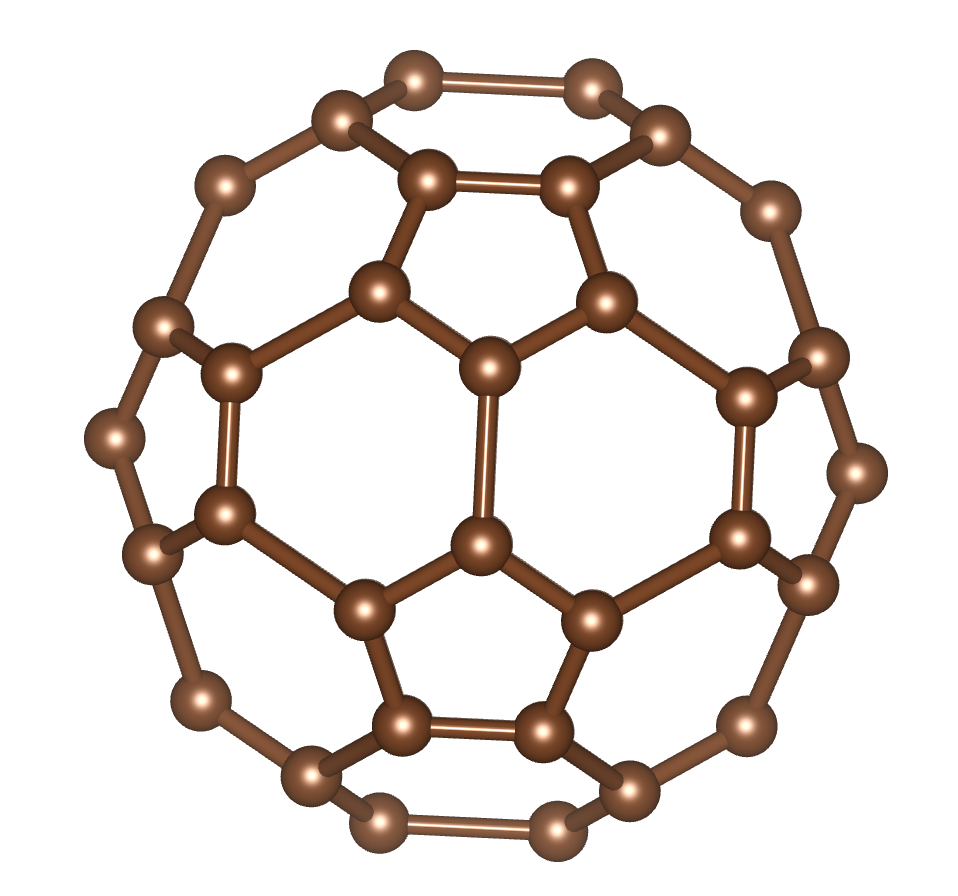
Note that this is a random process. So each time the structure is different.
3D crystals
By default, -d tag is 3, which means to generate 3D crystal. Below is a
quick example to generate a diamond like crystals for carbon.
$ pyxtal_main.py -e C -n 8 -s 227
Symmetry requested: 227(Fd-3m), generated: Fd-3m
Output to out/C8.cif
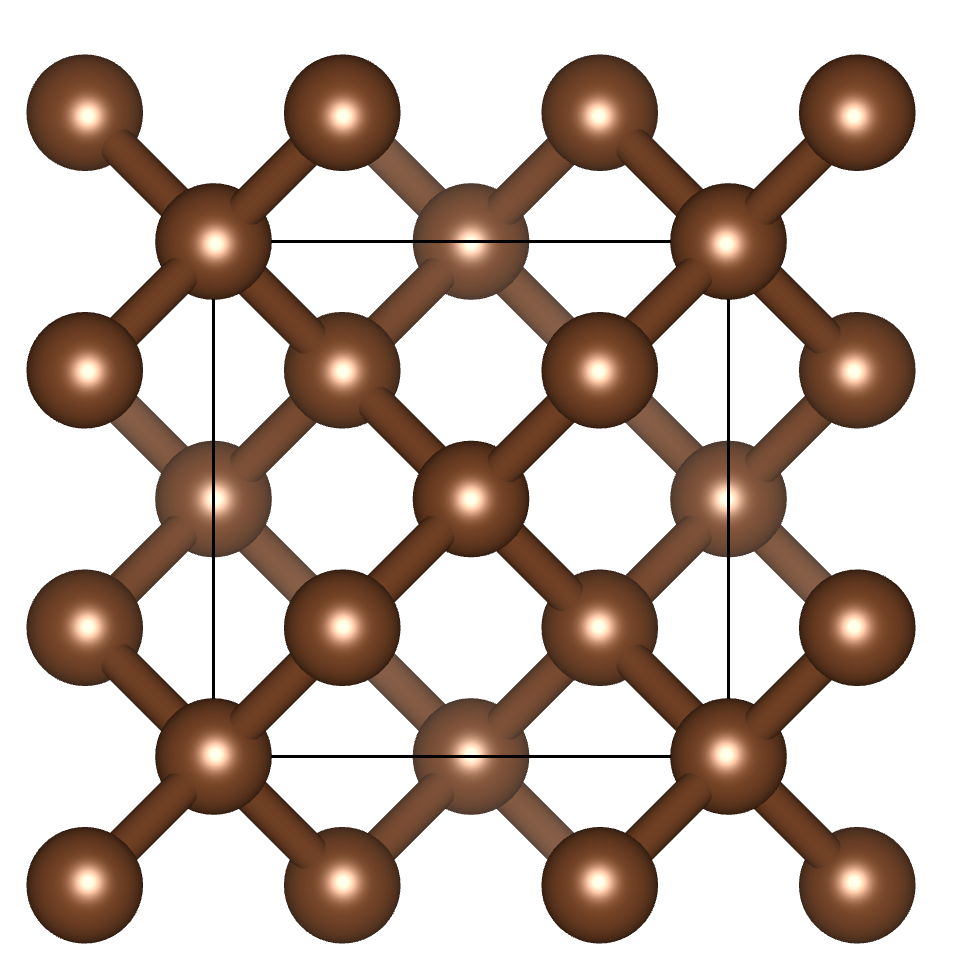
It is important to note that we specified 8 for -n tag, which means 8
carbon atoms in the conventional unit cell.
2D and 1D crystals
2D and 1D crystals need one more argument to specify the confinement. For 2D
crystal, the thickness needs to be provided through -t tag in Angstrom.
Below is an example fo generating a 2D MoS2 crystal.
$ pyxtal_main.py -e Mo,S -n 1,2 -s 77 -d 2 -t 2.4
Symmetry requested: 77(p6mm), generated: P6mm
Output to out/Mo1S2.cif
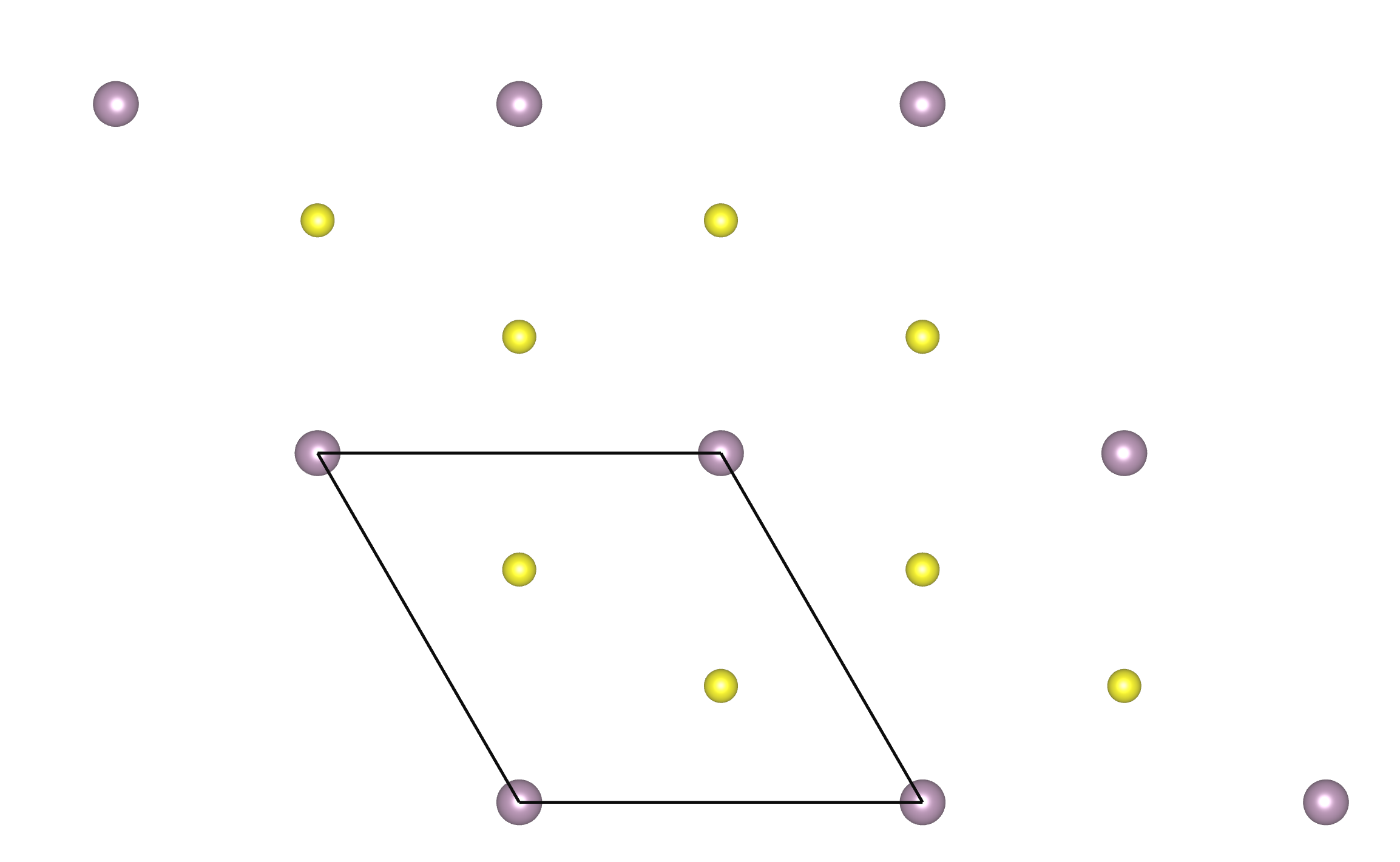
Molecular crystals occupying general Wyckoff positions
Below is an example to generate of random crystal for a famous drug molecule ROY.
$ pyxtal_main.py -m -e ROY -n 4 -s P2_12_12_1
Symmetry requested: 19 (P2_12_12_1), generated: P2_12_12_1, vol: 2895.37 A^3
Output to out/S4O8N12C48H36.cif
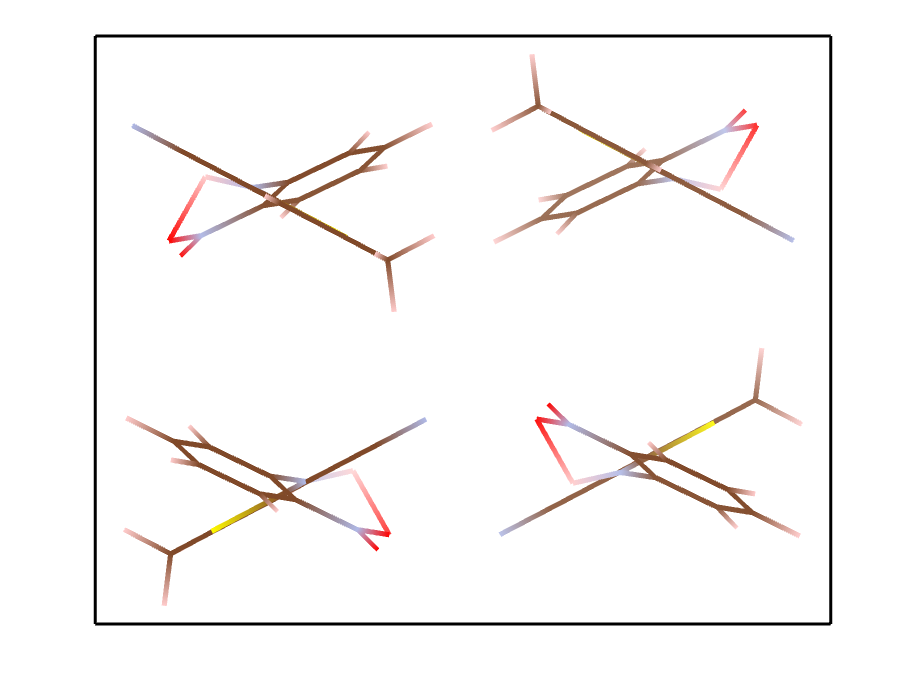
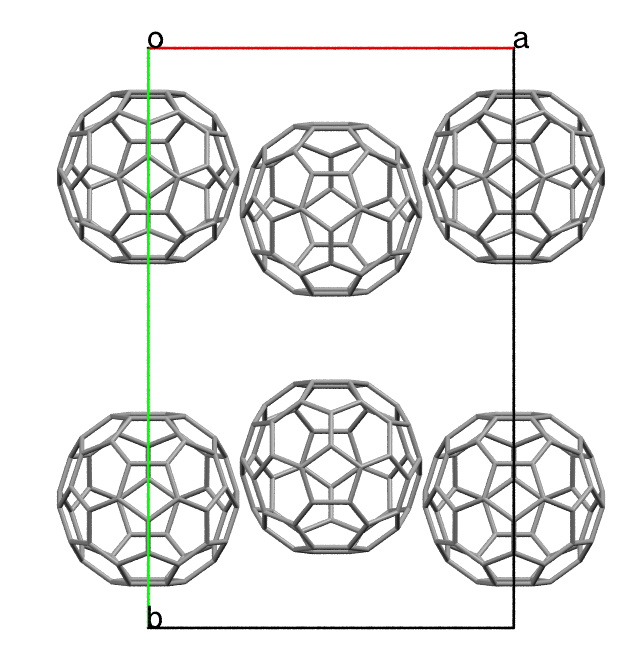
Molecular crystals occupying special Wyckoff positions
An import feature of PyXtal is that the program can automatically generate
molecular crystals occupying special Wyckoff positions. This is very useful for
molecules with high internal symmetry. During crystallization, these molecule
can occupy some special Wyckoff positions as long as the site symmetry is
compatible with the molecular symmetry. For instance, the space group Cmc_21
has 4 symmetry operations (mm2) in its primitive cell. However, we can still
generate a structure with 2 molecules for C60 by placing them to the special
Wyckoff position. This will be automatically processed by our
internal algorithm.
$ pyxtal_main.py -m -e C60 -n 2 -s 36
How to define the molecules?
Please ref to the section of Molecule.